Research
|
My current research interests focus on computational chemical and biological physics of condensed molecular systems, such as liquids, macromolecules and interfaces, using a wide variety of methods ranging from first principles molecular dynamics (MD), to force field-based and QM/MM-based MD. I am deeply interested in the development and the application of new methods, aiming, in particular, to the reliable and efficient description of structure, dynamics and vibrational properties. The code I mostly use are C2PK and CPMD. |
|
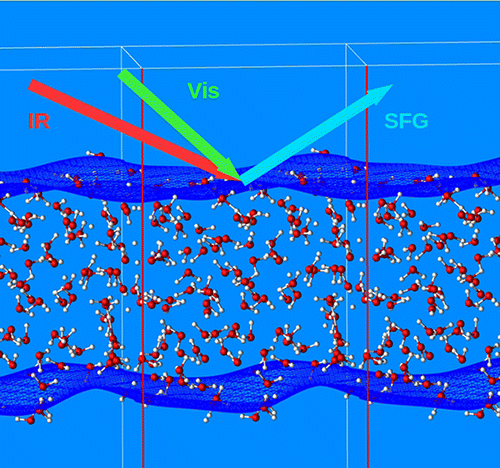
Vibrational spectroscopy of water interfaces We developed an expression for the calculation of the sum frequency generation spectra (SFG) of water interfaces that is based on the projection of the atomic velocities on the local normal modes. Our approach permits one to obtain the SFG signal from suitable velocity–velocity correlation functions, reducing the computational cost to that of the accumulation of a molecular dynamics trajectory, and therefore cutting the overhead costs associated with the explicit calculation of the dipole moment and polarizability tensor. Our method permits to interpret the peaks in the spectrum in terms of local modes, also including the bending region. Time-resolved SFG. What happens when extra vibrational energy is added to water? Using nonequilibrium molecular dynamics simulations, also including the full electronic structure, and novel descriptors, based on projected vibrational density of states, we are able to follow the flow of excess vibrational energy from the excited stretching and bending modes. |
|
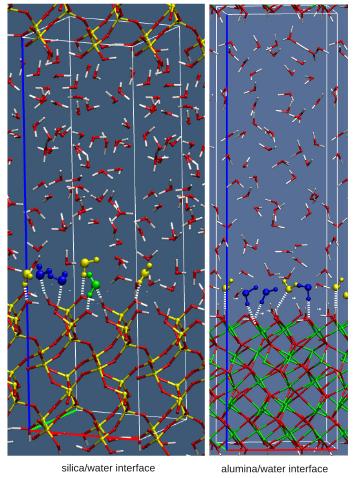
Solid/water interfaces structure and reactivity. We aim to a detailed understanding of the molecular behaviour of the different solidwater interfaces, using density functional theory based molecular dynamics (DFTMD) simulations, where a consistent treatment of the electronic structure of solvent and surface is provided. Our interest includes oxide water interfaces (such as silica, alumina, clays) as well as ionic salts/water interfaces, such as the fluoride/water interface. Environmentally relevant oxides-water interfaces The CaF2 /water interface |
|
|
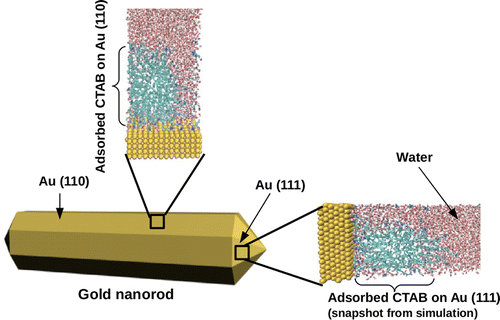
How the interface interactions influence the crystal growth. We investigate how the properties at the interface influence the crystal growth in a few selected example. In particular one topic is related to the understanding of the microscopic origin of the asymmetric growth mechanism in gold nanorods. The second topic is related to understanding how bio-polymers such as polyacrylate, poly-aspartate and poly-glutamate influence the crystalline phase, morphology and growth rate of calcium oxalate. Ab initio molecular dynamics study of the interactions of the water/mineral and water/polymer/mineral interfaces shed light on the biomineralization process and on the mechanisms responsible for its inhibition. Understanding the microscopic origin of gold nanoparticles anisotropic growth |
|
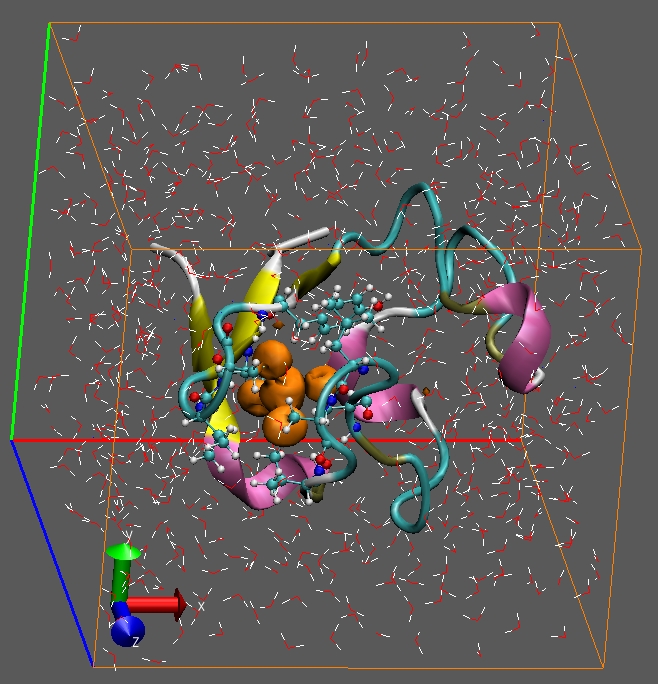
Computational electrochemistry in extended systems. We calculated the redox potential and reorganisation free energy for molecules in complex environments. We elucidated the role of the environment (solvent, protein scaffold) and its H-bond network on the electrochemical properties of several systems such as quinones and metalloproteins. |
|
Acidity constants and coupled proton/electron transfer.
Electron transfer is often coupled to proton transfer (PT) and this motivation has led us to develop a first principle simulation approach to compute acidity constants. The method is based on a half reaction scheme computing free energies of dissociation from the vertical energy gaps for insertion or removal of protons. |
Ab initio description of radicals in solution
QM/MM techniques: enzymatic catalysis and optical properties
Contact Dr. Sulpizi with any questions.