 |
[68]
Binder K, Hsu HP, Paul W
Eur. Phys. J. Special Topics
144: 154907 Apr 2
[67]
Hsu HP, Kremer K
Journal of Chemical Physics
144: 154907 Apr 2
[66]
Huang, AQ, Hsu HP, Bhattacharya A, Binder K
Journal of Chemical Physics
143: 243102
[65]
Egorov SA, Hsu HP, Milchev A, Binder K
Soft Matter
11: 2604
[64]
Hsu HP
Journal of Chemical Physics
141: 234901
[63]
Hsu HP
Journal of Chemical Physics
141: 164903 October 28, 2014
[62]
Journal of Chemical Physics
140: 204908 May 58, 2014
[59]
Pulling Single Adsorbed Bottle-Brush Polymers off a Flat Surface: A Monte Carlo Simulation
[58]
Semiflexible macromolecules with discrete bond angles confined in nanoslits: A Monte Carlo test of scaling concepts
46: 8017 September 23, 2013
Single semiflexible polymer chains confined in a planar slit geometry between parallel nonadsorbing repulsive walls a distance D apart are studied by Monte Carlo simulations of a lattice model, for the case of good solvent conditions. The polymers are modeled as self-avoiding walks on the simple cubic lattice, where every 90° kink requires a bending energy εb. For small qb = exp(−εb/kBT) the model has a large persistence length (given by ≈ 1/(4qb) in the bulk three-dimensional dilute solution, in units of the lattice spacing). Unlike the popular Kratky–Porod model of worm-like chains, this model takes both excluded volume into account and approximates the fact that bond angles between subsequent carbon–carbon bonds of real chains are (almost) restricted to large nonzero values, and the persistence length is controlled by torsional potentials. So the typical local conformation in the model is a straight sequence of (on average) lp bonds (roughly corresponding e.g. to an all-trans sequence of an alkane chain) followed by a 90° kink. While under weak confinement (D lp) the model (for very long chains) still is compatible with the Daoud–de Gennes scaling theory, for strong confinement (D ≤ lp) strong deviations from the predictions based on the Kratky–Porod model are found.
[57]
Semi-flexible polymer chains in quasi-one-dimensional confinement: a Monte Carlo study on the square lattice
9: 10512 June 26, 2013
Abstract:
Single semi-flexible polymer chains are modeled as self-avoiding walks (SAWs) on the square lattice with every 90° kink requiring an energy εb. While for εb = 0 this is the ordinary SAW, varying the parameter qb = exp(−εb/kBT) allows the variation of the effective persistence length
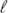 p over about
two decades. Using the pruned-enriched Rosenbluth method
(PERM), chain lengths up to about N = 105
steps can be studied. In previous work it has already been
shown that for contour lengths L = Nb (the bond
length b is the
lattice spacing) of order p a smooth
crossover from rods to two-dimensional self-avoiding walks
occurs, with radii R ∝ p1/4L3/4,
the Gaussian regime predicted by the Kratky–Porod model for
worm-like chains being completely absent. In the present
study, confinement of such chains in strips of width D
is considered, varying D from 4 to 320 lattice
spacings. It is shown that for narrow strips (D <
p) the
effective persistence length of the chains (in the direction
parallel to the confining boundaries) scales like p2/D,
and R‖ ∝ L (with
a pre-factor of order unity). For very wide strips, D
≫ p, the
two-dimensional SAW behavior prevails for chain lengths up to
Lcross ∝ p(D/p)4/3,
while for L ≫ Lcross
the chain is a string of blobs of diameter D, i.e.
R‖ ∝ L(p/D)1/3.
In the regime D < p, the chain is
a sequence of straight sequences with length of the order p2/D
parallel to the boundary, separated by sequences with length
< D perpendicular to the boundary; thus Odijk's
deflection length plays no role for discrete bond angles.
p over about
two decades. Using the pruned-enriched Rosenbluth method
(PERM), chain lengths up to about N = 105
steps can be studied. In previous work it has already been
shown that for contour lengths L = Nb (the bond
length b is the
lattice spacing) of order p a smooth
crossover from rods to two-dimensional self-avoiding walks
occurs, with radii R ∝ p1/4L3/4,
the Gaussian regime predicted by the Kratky–Porod model for
worm-like chains being completely absent. In the present
study, confinement of such chains in strips of width D
is considered, varying D from 4 to 320 lattice
spacings. It is shown that for narrow strips (D <
p) the
effective persistence length of the chains (in the direction
parallel to the confining boundaries) scales like p2/D,
and R‖ ∝ L (with
a pre-factor of order unity). For very wide strips, D
≫ p, the
two-dimensional SAW behavior prevails for chain lengths up to
Lcross ∝ p(D/p)4/3,
while for L ≫ Lcross
the chain is a string of blobs of diameter D, i.e.
R‖ ∝ L(p/D)1/3.
In the regime D < p, the chain is
a sequence of straight sequences with length of the order p2/D
parallel to the boundary, separated by sequences with length
< D perpendicular to the boundary; thus Odijk's
deflection length plays no role for discrete bond angles.Full Text: Soft Matter
[56]
Estimation of persistence lengths of semiflexible polymers: Insight from simulations
55: 39 September 1, 2013
Abstract:
The persistence length of macromolecules is one of their basic characteristics, describing their intrinsic local stiffness. However, it is difficult to extract this length from physical properties of the polymers, different recipes may give answers that disagree with each other. Monte Carlo simulations are used to elucidate this problem, giving a comparative discussion of two lattice models, the self-avoiding walk model extended by a bond bending energy, and bottle-brush polymers described by the bond fluctuation model. The conditions are discussed under which a description of such macromolecules by Kratky-Porod worm-like chains holds, and the question to what extent the persistence length depends on external conditions (such as solvent quality) is considered. The scattering function of semiflexible polymers is discussed in detail, a comparison to various analytic treatments is given, and an outlook to experimental work is presented.
[55]
87: 022604 Febuary 27, 2013
Abstract:
The coil-bridge transition in a self-avoiding lattice chain with one end fixed at height H above the attractive planar surface is investigated by theory and Monte Carlo simulation. We focus on the details of the first-order phase transition between the coil state at large height H ⩾ H tr and a bridge state at H ⩽ H tr , where H tr corresponds to the coil-bridge transition point. The equilibrium properties of the chain were calculated using the Monte Carlo pruned-enriched Rosenbluth method in the moderate adsorption regime at (H/Na) tr ⩽ 0.27 where N is the number of monomer units of linear size a. An analytical theory of the coil-bridge transition for lattice chains with excluded volume interactions is presented in this regime. The theory provides an excellent quantitative description of numerical results at all heights, 10 ⩽ H/a ⩽ 320 and all chain lengths 40 < N < 2560 without free fitting parameters. A simple theory taking into account the effect of finite extensibility of the lattice chain in the strong adsorption regime at (H/Na) tr ⩾ 0.5 is presented. We discuss some unconventional properties of the coil-bridge transition: the absence of phase coexistence, two micro-phases involved in the bridge state, and abnormal behavior in the microcanonical ensemble.
Polymers grafted with one chain end to an impenetrable flat hard wall which attracts the monomers with a short-range adsorption potential (of strength ε) are studied by large scale Monte Carlo simulations, using the pruned–enriched Rosenbluth method (PERM). Chain lengths up to N = 25600 steps are considered, and the intrinsic flexibility of the chain is varied via an energy penalty for nonzero bond angles, εb. Choosing qb = exp(−εb/kBT) in the range from qb = 1 (fully flexible chains) to qb = 0.005 (rather stiff chains with a persistence length of about
 lattice spacings), the adsorption transition is
found to vary from about ε/kBTc
≈ 0.286 to ε/kBTc
≈ 0.011, confirming the theoretical expectation that
lattice spacings), the adsorption transition is
found to vary from about ε/kBTc
≈ 0.286 to ε/kBTc
≈ 0.011, confirming the theoretical expectation that  for large
for large  . The simulation data are compatible with a
continuous adsorption transition for all finite values of
. The simulation data are compatible with a
continuous adsorption transition for all finite values of  , while in the rigid rod limit (
, while in the rigid rod limit ( ) a first order transition seems to emerge. Scaling
predictions and blob concepts on the structure of weakly
adsorbed semiflexible polymers absorbed at interfaces are
briefly discussed.
) a first order transition seems to emerge. Scaling
predictions and blob concepts on the structure of weakly
adsorbed semiflexible polymers absorbed at interfaces are
briefly discussed.[54]
87: 022604 Febuary 27, 2013
Abstract:
We investigate the effects of the range of adsorption potential on the equilibrium behavior of a single polymer chain end-attached to a solid surface. The exact analytical theory for ideal lattice chains interacting with a planar surface via a box potential of depth U and width W is presented and compared to continuum model results and to Monte Carlo (MC) simulations using the pruned-enriched Rosenbluth method for self-avoiding chains on a simple cubic lattice. We show that the critical value U-c corresponding to the adsorption transition scales as W-1/nu,where the exponent nu = 1/2 for ideal chains and nu approximate to 3/5 for self-avoiding walks. Lattice corrections for finite W are incorporated in the analytical prediction of the ideal chain theory U-c approximate to (pi(2)/24)(W + 1/2)(-2) and in the best-fit equation for the MC simulation data U-c = 0.585(W + 1/2)(-5/3). Tail, loop, and train distributions at the critical point are evaluated by MC simulations for 1 <= W <= 10 and compared to analytical results for ideal chains and with scaling theory predictions. The behavior of a self-avoiding chain is remarkably close to that of an ideal chain in several aspects. We demonstrate that the bound fraction theta and the related properties of finite ideal and self-avoiding chains can be presented in a universal reduced form: theta(N, U, W) = theta(NUc, U/U-c). By utilizing precise estimations of the critical points we investigate the chain length dependence of the ratio of the normal and lateral components of the gyration radius. Contrary to common expectations this ratio attains a limiting universal value < R-g perpendicular to(2)>/< R-g parallel to(2)> = 0.320 +/- 0.003 only at N similar to 5000. Finite-N corrections for this ratio turn out to be of the opposite sign for W = 1 and for W >= 2. We also study the N dependence of the apparent crossover exponent phi(eff)(N). Strong corrections to scaling of order N-0.5 are observed, and the extrapolated value phi = 0.483 +/- 0.003 is found for all values of W. The strong correction to scaling effects found here explain why for smaller values of N, as used in most previous work, misleadingly large values of phi(eff)(N) were identified as the asymptotic value for the crossover exponent.
[53]
137: 174902 November 6, 2012
Abstract:
Using the pruned-enriched Rosenbluth Monte Carlo algorithm, the scattering functions of semiflexible macromolecules in dilute solution under good solvent conditions are estimated both in d = 2 and d = 3 dimensions, considering also the effect of stretching forces. Using self-avoiding walks of up to N = 25 600 steps on the square and simple cubic lattices, variable chain stiffness is modeled by introducing an energy penalty εb for chain bending; varying qb = exp (−εb/kBT) from qb = 1 (completely flexible chains) to qb = 0.005, the persistence length can be varied over two orders of magnitude. For unstretched semiflexible chains, we test the applicability of the Kratky-Porod worm-like chain model to describe the scattering function and discuss methods for extracting persistence length estimates from scattering. While in d = 2 the direct crossover from rod-like chains to self-avoiding walks invalidates the Kratky-Porod description, it holds in d = 3 for stiff chains if the number of Kuhn segments nK does not exceed a limiting value nK* (which depends on the persistence length). For stretched chains, the Pincus blob size enters as a further characteristic length scale. The anisotropy of the scattering is well described by the modified Debye function, if the actual observed chain extension ⟨X⟩ (end-to-end distance in the direction of the force) as well as the corresponding longitudinal and transverse linear dimensions ⟨X2⟩ − ⟨X⟩2, 〈Rg,⊥2〉 are used.
[52]
136: 024901 January 14, 2012
Abstract:
Semiflexible macromolecules in dilute solution under very good solvent conditions are modeled by self-avoiding walks on the simple cubic lattice (d = 3 dimensions) and square lattice (d = 2 dimensions), varying chain stiffness by an energy penalty εb for chain bending. In the absence of excluded volume interactions, the persistence length ℓp of the polymers would then simply be ℓp = ℓb(2d−2)−1qb−1 with qb = exp (−εb/kBT), the bond length ℓb being the lattice spacing, and kBT is the thermal energy. Using Monte Carlo simulations applying the pruned-enriched Rosenbluth method (PERM), both qb and the chain length N are varied over a wide range (0.005 ⩽ qb ⩽ 1, N ⩽ 50 000), and also a stretching force f is applied to one chain end (fixing the other end at the origin). In the absence of this force, in d = 2 a single crossover from rod-like behavior (for contour lengths less than ℓp) to swollen coils occurs, invalidating the Kratky-Porod model, while in d = 3 a double crossover occurs, from rods to Gaussian coils (as implied by the Kratky-Porod model) and then to coils that are swollen due to the excluded volume interaction. If the stretching force is applied, excluded volume interactions matter for the force versus extension relation irrespective of chain stiffness in d = 2, while theories based on the Kratky-Porod model are found to work in d = 3 for stiff chains in an intermediate regime of chain extensions. While for qb ≪ 1 in this model a persistence length can be estimated from the initial decay of bond-orientational correlations, it is argued that this is not possible for more complex wormlike chains (e.g., bottle-brush polymers). Consequences for the proper interpretation of experiments are briefly discussed.
[51]
Computer simulation of bottle-brush polymers with flexible backbone: Good solvent versus theta solvent conditions
PE, Hsu HP, Paul W, Binder K
Journal of Chemical Physics
135: 164903 October 31, 2011
[50]
Scaling behaviour of lattice animals at the upper critical dimension
von Ferber C, Foster D, Hsu HP, Kenna R
The European Physical Journal B
83: 245-249 15 September, 2011
Full Text: EPJB or cond-mat
[49]
Breakdown of the Kratky-Porod Wormlike Chain Model for Semiflexible Polymers in Two Dimensions
Hsu HP, Paul W, Binder K
Europhysics Letters
95: 68004 September 6, 2011
[48]
A Review of Monte Carlo Simulations of Polymers with PERM
Hsu HP, Grassberger P
Journal of Statistical Physics
144: 597-637 July 20, 2011
[47]
Structure of Bottle brush Polymers on Surfaces: Weak versus Strong Adsorption
Hsu HP, Paul W, Binder K
Journal of Physical Chemistry B
DOI: 10.1021/jp204006z
 40% of the monomers being
bound to the surface, forcing the bottle brush into an
almost 2D conformation) are studied. We focus on the
scaling of the total linear dimensions of the
cylindrical brush with both chain lengths N and Nb, demonstrating a crossover
from rod-like behavior (for not very large Nb) to the scaling of 2D
self-avoiding walks. Despite the fact that snapshot
pictures suggest a “worm-like” picture as a
coarse-grained description of such cylindrical brushes,
the Kratky–Porod worm-like chain model fails because
there is no regime where Gaussian statistics applies. We
compare the stiffness (orientational correlations of
backbone bonds, persistence length estimates, etc.) of
the adsorbed bottle brush polymers with their
corresponding 3D nonadsorbed counterparts. Consequences
for the discussion of pertinent experiments are briefly
discussed.
40% of the monomers being
bound to the surface, forcing the bottle brush into an
almost 2D conformation) are studied. We focus on the
scaling of the total linear dimensions of the
cylindrical brush with both chain lengths N and Nb, demonstrating a crossover
from rod-like behavior (for not very large Nb) to the scaling of 2D
self-avoiding walks. Despite the fact that snapshot
pictures suggest a “worm-like” picture as a
coarse-grained description of such cylindrical brushes,
the Kratky–Porod worm-like chain model fails because
there is no regime where Gaussian statistics applies. We
compare the stiffness (orientational correlations of
backbone bonds, persistence length estimates, etc.) of
the adsorbed bottle brush polymers with their
corresponding 3D nonadsorbed counterparts. Consequences
for the discussion of pertinent experiments are briefly
discussed.Full Text: J. Phys. Chem. B
[46]
Hsu HP, Paul W
Computer Physics Communications
182: 2115-2121 October 25, 2011
[45]
Hsu HP
Physics Procedia
15: 44-53 July 22, 2011
Full Text: Physics Procedia or cond-mat
[44]
Hsu HP, Paul W, Binder K
Macromolecular Theory and Simulations
20: 510-525 August 25, 2011
Bottle-brush polymers contain a long flexible macromolecule as a backbone to which flexible side chains are grafted. Through the choice of the grafting density and the length of the side chains the local stiffness of this cylindrical molecular brush can be controlled, but a quantitative understanding of these phenomena is lacking. Monte Carlo simulation results are presented and discussed which address this issue, extracting mesoscopic length scales (such as the cross-sectional radius, persistence length, and contour length of these objects). Large-scale simulations of the bond fluctuation model are combined with simulations of the simple self-avoiding walk (SAW) model with flexibility controlled by a bond-angle potential, using the pruned-enriched Rosenbluth algorithm. It is shown that under good solvent conditions the bottle-brush polymers never display a pre-asymptotic Gaussian regime that would be described by the Kratky–Porod worm-like chain model, unlike the semiflexible SAW model. Implications of these results for the proper interpretation of experiments are discussed.
[43]
Hsu HP, Paul W, Binder K
EUROPHYS LETTER
92: 28003 (6 pages) 15 November 2010
When the local intrinsic stiffness of a polymer chain varies over a wide range, one can observe both a crossover from rigid-rod–like behavior to (almost) Gaussian random coils and a further crossover towards self-avoiding walks in good solvents. Using the pruned-enriched Rosenbluth method (PERM) to study self-avoiding walks of up to Nb=50000 steps and variable flexibility, the applicability of the Kratky-Porod model is tested. Evidence for non-exponential decay of the bond-orientational correlations ⟨cos θ(s)⟩ for large distances s along the chain contour is presented, irrespective of chain stiffness. For bottle-brush polymers on the other hand, where experimentally stiffness is varied via the length of side-chains, it is shown that these cylindrical brushes (with flexible backbones) are not described by the Kratky-Porod worm-like chain model, since their persistence length is (roughly) proportional to their cross-sectional radius, for all conditions of practical interest.
[42]
Hsu HP, Paul W, Binder K
JOURNAL OF CHEMICAL PHYSICS
133: 134902 (14 pages) 7 October 2010
The adsorption of a bottle-brush polymer end-grafted with one chain end of its backbone to a flat substrate surface is studied by Monte Carlo simulation of a coarse-grained model, that previously has been characterized in the bulk, assuming a dilute solution under good solvent conditions. Applying the bond fluctuation model on the simple cubic lattice, we vary the backbone chain length Nb from Nb = 67 to Nb = 259 effective monomeric units, the side chain length N from N = 6 to N = 48, and set the grafting density to σ = 1, i.e., parameters that correspond well to the experimentally accessible range. When the adsorption energy strength ϵ is varied, we find that the adsorption transition (which becomes well-defined in the limit Nb→∞, for arbitrary finite N) roughly occurs at the same value ϵc as for ordinary linear chains (N = 0), at least within our statistical errors. Mean square end-to-end distances and gyration radii of the side chains are obtained, as well as the monomer density profile in the direction perpendicular to the adsorbing surface. We show that for longer side chains the adsorption of bottle-brushes is a two-step process, the decrease of the perpendicular linear dimension of side chains with adsorption energy strength can even be nonmonotonic. Also, the behavior of the static structure factor S(q) is analyzed, evidence for a quasi-two-dimensional scaling is presented, and consequences for the interpretation of experiments are discussed.
[41]
Bolinger D, Sulkowska, JI, Hsu HP, Mirny LA, Kardar M, Onuchic JN, Virnau P
PLOS COMPUTATIONAL BIOLOGY
6(4): e1000731 APRIL 2010
Protein knots, mostly regarded as intriguing oddities, are gradually being recognized as significant structural motifs. Seven distinctly knotted folds have already been identified. It is by and large unclear how these exceptional structures actually fold, and only recently, experiments and simulations have begun to shed some light on this issue. In checking the new protein structures submitted to the Protein Data Bank, we encountered the most complex and the smallest knots to date: A recently uncovered alpha-haloacid dehalogenase structure contains a knot with six crossings, a so-called Stevedore knot, in a projection onto a plane. The smallest protein knot is present in an as yet unclassified protein fragment that consists of only 92 amino acids. The topological complexity of the Stevedore knot presents a puzzle as to how it could possibly fold. To unravel this enigma, we performed folding simulations with a structure-based coarse-grained model and uncovered a possible mechanism by which the knot forms in a single loop flip.
[40]
Hsu HP, Paul W, Binder K
MACROMOLECULES
43(6): 3094-3102 MARICH 23 2010
On the basis of extensive Monte Carlo simulations of lattice models for linear chains under good and Theta solvents conditions, and for bottle-brush polymers under good solvent conditions, different methods to estimate the persistence lengths of these polymers are applied and compared to each other. While for chain molecules at the Theta point standard textbook definitions of the persistence length yield consistent results, under good solvent conditions the persistence length (according to its standard definitions) diverges when the chain length of the macromolecules tends to infinity. Accurate simulation results for chain lengths up to N-b = 6400 allow us to verify the theoretically predicted power laws for the decay of the bond orientational correlation function. For the case of bottle-brush polymers, this dependence of "the" persistence length on the backbone chain length obscures the dependence on the side chain length, that is controversially discussed in the literature. Alternative definitions Or a persistence length that do not suffer from this problem, based on the total linear dimension of the chain or on the scattering function via the so-called "Holtzer plateau" are studied as well. We show that the backbone contour length of the bottle-brush needs to be very large (about 100 persistence lengths in typical cases) to reach the asymptotic limit where the bottle-brush satisfies the self-avoiding walk statistics, and where a well-defined persistence length can be extracted. An outlook to pertinent experimental work is given.
[39]
Hsu HP, Paul W, Rathheber S, Binder K
MACROMOLECULES
43(3): 1592-1601 JANUARY 7 2010
Extensive Monte Carlo simulations are presented for bottle-brush polymers under good solvent conditions, using the bond fluctuation model on the simple cubic lattice. Varying the backbone length (from Nb = 67 to Nb = 259 effective monomers) as well as the side chain length (from N = 6 to N = 48), for a physically reasonable grafting density of one chain per backbone monomer, we find that the structure factor describing the total scattering from the bottle-brush provides an almost perfect match for some combinations of (Nb, N) to experimental data of Rathgeber et al. [ J. Chem. Phys. 2005, 122, 124904], when we adjust the length scale of the simulation to reproduce the experimental gyration radius of the bottle-brush. While in the experiment other length scales (gyration radius of side chains, backbone persistence length, scale characterizing the radial monomer density profile in the plane normal to the backbone) can be extracted only via fitting to a complicated and approximate theoretical expression derived by Pedersen and Schurtenberger, all these properties can be extracted from the simulation directly. In this way, quantitatively more reliable estimates for the persistence length and side chain gyration radius of the experimental systems can be extracted. In particular, we show that the popular assumption of a Gaussian radial monomer density profile is inaccurate, in the very good solvent regime studied by the simulation, and show that alternative forms based on scaling theory work better. We also show that the persistence length of the bottle brush in the simulation depends systematically on the backbone length and not only on the side chain length. For the cases where an explicit comparison with the experimental results (based on their evaluation within the Pedersen−Schurtenberger model) is possible, simulation and experiment are consistent with each other and some of the (rather minor) differences between simulation and experiment can be attributed to the weaker strength of excluded volume in the latter. Thus, we show that by suitable mapping between simulation and experiment on length scales of the local concentration fluctuations (here <2 nm) the analysis of experimental data can be systematically refined.
[36]
Hsu HP, Binder K, Paul W
PHYSICAL REVIEW LETT
103(4): 198301 (4 pages), NOV 2009
[35]
Structure of
bottle-brush polymers in solution: A Monte Carlo test of
models for the scattering function
Hsu HP, Paul W, Binder
K
JOURNAL OF CHEMICAL PHYSICS
129: 204904-1-204904-11
Nov 28 2008
Full Text: JCP or cond-mat
[34]
Kenna, R.; Hsu, HP, von Ferber, C
JOURNAL OF STATISTICAL MECHANICS-THEORY AND EXPERIMENT
Art. No. L10002 OCT 2008
Abstract:
For continuous phase transitions characterized by power-law divergences, Fisher renormalization prescribes how to obtain the critical exponents for a system under constraint from their ideal counterparts. In statistical mechanics, such ideal behaviour at phase transitions is frequently modified by multiplicative logarithmic corrections. Here, Fisher renormalization for the exponents of these logarithms is developed in a general manner. As for the leading exponents, Fisher renormalization at the logarithmic level is seen to be involutory and the renormalized exponents obey the same scaling relations as their ideal analogues. The scheme is tested in lattice animals and the Yang Lee problem at their upper critical dimensions, where predictions for logarithmic corrections are made.
Full Text: JStatM or cond-mat
[33]
Escape transition of a polymer
chain from a nanotube: How to avoid spurious results by
use of the force-biased pruned-enriched Rosenbluth
algorithm
Hsu HP, Binder K, Klushin LI, Skvortsov AM
PHYSICAL REVIEW E
78 (4): 041803-1 - 041803-11, OCT 2008
A polymer chain containing N monomers confined in a finite cylindrical tube of diameter D grafted at a distance L from the open end of the tube may undergo a rather abrupt transition, where part of the chain escapes from the tube to form a "crownlike" coil outside of the tube. When this problem is studied by Monte Carlo simulation of self-avoiding walks on the simple cubic lattice applying a cylindrical confinement and using the standard pruned-enriched Rosenbluth method (PERM), one obtains spurious results, however, with increasing chain length the transition gets weaker and weaker, due to insufficient sampling of the "escaped" states, as a detailed analysis shows. In order to solve this problem, a new variant of a biased sequential sampling algorithm with resampling is proposed, force-biased PERM: the difficulty of sampling both phases in the region of the first order transition with the correct weights is treated by applying a force at the free end pulling it out of the tube. Different strengths of this force need to be used and reweighting techniques are applied. Using rather long chains (up to N=18 000) and wide tubes (up to D=29 lattice spacings), the free energy of the chain, its end-to-end distance, the number of "imprisoned" monomers can be estimated, as well as the order parameter and its distribution. It is suggested that this algorithm should be useful for other problems involving state changes of polymers, where the different states belong to rather disjunct "valleys" in the phase space of the system.
Full Text: PRE or cond-mat
[32]
Dragging a
polymer chain into a nanotube and subsequent release
Klushin
LI, Skvortsov AM, Hsu HP,
Binder K
MACROMOLECULES
41(15): 5890-5898 JULY 2
2008
We present a scaling theory and Monte Carlo (MC) simulation results for a flexible polymer chain slowly dragged by one end into a nanotube. We also describe the situation when the completely confined chain is released and gradually leaves the tube. MC simulations were performed for a self-avoiding lattice model with a biased chain growth algorithm, the pruned-enriched Rosenbluth method (PERM). The nanotube is a long channel opened at one end and its diameter D is much smaller than the size of the polymer coil in solution. We analyze the following characteristics as functions of the chain end position x inside the tube: the free energy of confinement, the average end-to-end distance, the average number of segments imprisoned in the tube, and the average stretching of the confined part of the chain for various values of D and for the number of repeat units in the chain, N. We show that when the chain end is dragged by a certain critical distance x* into the tube, the polymer undergoes a first-order phase transition whereby the remaining free tail is abruptly sucked into the tube. This is accompanied by jumps in the average size, the number of imprisoned segments, and the average stretching parameter. The critical distance scales as x* similar to ND1-1/v. The transition takes place when approximately 3/4 of the chain units are dragged into the tube. The theory presented is based on constructing the Landau free energy as a function of an order parameter that provides a complete description of equilibrium and metastable states. We argue that if the trapped chain is released with all monomers allowed to fluctuate, the reverse process in which the chain leaves the confinement occurs smoothly without any jumps. Finally, we apply the theory to estimate the lifetime of confined DNA in metastable states in nanotubes.
[31]
Adsorption
of
Multi-block
and Random Copolymer on a Solid Surface: Critical
Behavior and Phase Diagram
Bhattacharya S, Hsu HP, Milchev,
Rostiashvili VG, Vilgis TA
MACROMOLECULES
41(8): 2920-2930 MARCH 29
2008
[30]
One- and two-component
bottle-brush polymers: simulations compared to
theoretical predictions
Hsu HP, Paul W, Binder
K
MACROMOLECULAR THEORY AND SIMULATIONS
16 (7): 660-689 SEP 25
2007
Full Text: MTS
[28]
What is the order of the
two-dimensional polymer escape transition?
Hsu HP, Binder K, Klushin LI, Skvortsov AM
PHYSICAL REVIEW E
76 (2): 021108-1 - 021108-14, AUG 2007
An end-grafted flexible polymer chain in three-dimensional space between two pistons undergoes an abrupt transition from a confined coil to a flowerlike conformation when the number of monomers in the chain, N, reaches a critical value. In two-dimensional (2D) geometry, excluded-volume interactions between monomers of a chain confined inside a strip of finite length 2L transform the coil conformation into a linear string of blobs. However, the blob picture raises questions about the nature of this escape transition. To check theoretical predictions based on the blob picture we study 2D single-polymer chains with excluded-volume interactions and with one end grafted in the middle of a strip of length 2L and width H by simulating self-avoiding walks on a square lattice with the pruned-enriched Rosenbluth method. We estimate the free energy, the end-to-end distance, the number of imprisoned monomers, the order parameter, and its distribution. It is shown that in the thermodynamic limit of large N and L but finite L/N, there is a small but finite jump in several average characteristics, including the order parameter. We also present a theoretical description based on the Landau free energy approach, which is in good agreement with the simulation results. Both simulation results and the analytical theory indicate that the 2D escape transition is a weak first-order phase transition.
Full Text: PRE or cond-mat
[27]
Hsu HP, Grassberger P
77 (1): 18003-P1-18003-P4 JAN 2007
We study a single flexible chain molecule grafted to a membrane which has pores of size slightly larger than the monomer size. On both sides of the membrane there is the same solvent. When this solvent is good, i.e. when the polymer is described by a self-avoiding walk, it can fairly easily penetrate the membrane, so that the average number of membrane crossings tends, for chain length N→∞, to a positive constant. The average numbers of monomers on either side of the membrane diverges in this limit, although their ratio becomes infinite. For a poor solvent, in contrast, the entire polymer is located, for large N, on one side of the membrane. For good and for theta solvents (ideal polymers) we find scaling laws, whose exponents can in the latter case be easily understood from the behaviour of random walks.
Full Text: EPL or cond-mat
[26]
76 (3): 526-532 NOV 2006
A lattice model for a symmetrical copolymer "bottle brush" molecule, where two types (A, B) of flexible side chains are grafted with one chain end to a rigid backbone, is studied by a variant of the pruned-enriched Rosenbluth method (PERM), allowing for simultaneous growth of all side chains in the Monte Carlo sampling. Choosing repulsive binary interactions between unlike monomers and varying the solvent quality, it is found that phase separation into an A-rich part of the cylindrical molecule and a B-rich part can occur only locally. Lon-grange order (in the direction of the backbone) does not occur, and hence the transition from the randomly mixed state of the bottle brush to the phase separated structure is strongly rounded, in contrast to corresponding mean-field predictions. This lack of a phase transition can be understood from an analogy with spin models in one space dimension. We predict that the range of microphase separation along the bottle brush backbone can be controlled on the nanoscale by varying the solvent quality.
Full Text: EPL or cond-mat
[25]
wedges, cones, and branch points of Riemann surfaces
Hsu HP, Nadler W, Grassberger P
PHYSICAL REVIEW E
71 (6): Art. No. 065104 Part 2, JUN 2005
degrees, or can have boundaries, generalizing wedges. We find conformal invariance behavior, $\theta \sim 1/\alpha$, only for small angles ($\alpha \ll 2\pi$), while $\theta \approx const -\alpha/2\pi$ for $\alpha \gg 2\pi$. These scalings hold both for wedges and cones. A heuristic (non-conformal) argument for the behavior at large $\alpha$ is given, and comparison is made with critical percolation.
Hsu HP, Grassberger P
JOURNAL OF STATISTICAL MECHANICS-THEORY AND EXPERIMENT
Art. No. P06003 JUN 2005
[23]
Hsu HP, Grassberger P
JOURNAL OF STATISTICAL MECHANICS-THEORY AND EXPERIMENT
Art. No. P01007 JAN 2005
Abstract:
Full Text: JStatM or cond-mat
[22]
Hsu HP, Nadler W,Grassberger P
JOURNAL OF PHYSICS A-MATHEMATICAL AND GENERAL
38 (4): 775-806 JAN 28 2005
Abstract:
Full Text: JPA or cond-mat
[20]
Hsu HP, Grassberger P
EUROPHYSICS LETTERS
66 (6): 874-880 JUN 2004
Abstract:
Full Text: EPL or cond-mat
[19]
Hsu HP, Nadler W, Grassberger P
MACROMOLECULES
37 (12): 4658-4663 JUN 15 2004
Abstract:
Full Text: MACROMOLECULES
[17]
Berg BA, Hsu HP
PHYSICAL REVIEW E
Abstract:
Full Text: PRE or cond-mat
Hsu HP, Grassberger P
JOURNAL OF CHEMICAL PHYSICS
Full Text: JCP or cond-mat
Hsu HP, Grassberger P
EUROPEAN PHYSICAL JOURNAL B
Abstract:
Full Text: EPJB or cond-mat
[13]
Hsu HP, Mehra V, Grassberger P
PHYSICAL REVIEW E 68
(2): Art. No. 037703 2003
Full Text: PRE or cond-mat
[12]
Hsu HP, Mehra V, Nadler W, Grassberger P
PHYSICAL REVIEW E
[11]
Hsu HP, Mehra V, Nadler W, Grassberger P
JOURNAL OF CHEMICAL PHYSICS
Hsu HP, Grassberger P
JOURNAL OF PHYSICS A-MATHEMATICAL AND GENERAL